
SOTYKTU 6 mg, comprimé pelliculé, boîte de 2 plaquettes calendaires de 14
Dernière révision : 03/07/2024
Taux de TVA : 2.1%
Prix de vente : 618,03 €
Taux remboursement SS : 30%
Base remboursement SS : 618,03 €
Laboratoire exploitant : BRISTOL-MYERS SQUIBB
Source :

SOTYKTU est indiqué dans le traitement du psoriasis en plaques modéré à sévère chez les adultes éligibles à un traitement systémique.
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients. Infections actives cliniquement importantes (par exemple : tuberculose active, voir rubrique Mises en garde spéciales et précautions d'emploi).
Infections
Le deucravacitinib peut augmenter le risque d'infections (voir rubrique Effets indésirables).
Le traitement par deucravacitinib ne doit pas être instauré chez les patients présentant une infection active importante sur le plan clinique tant que l'infection n'est pas résolue ou correctement traitée (voir rubrique Contre-indications). L'utilisation du deucravacitinib chez les patients présentant une infection chronique ou un antécédent d'infection récurrente doit être envisagée avec précaution.
Il convient d'indiquer aux patients traités par deucravacitinib de demander un avis médical en cas d'apparition de signes ou symptômes évoquant une infection. Si un patient développe une infection importante sur le plan clinique ou ne répond pas au traitement standard, le patient doit être surveillé attentivement et le deucravacitinib ne doit pas être administré tant que l'infection n'est pas résolue.
Évaluation préalable au traitement pour la tuberculose
Avant de débuter un traitement par deucravacitinib, les patients doivent effectuer un test de dépistage de l'infection par la tuberculose (TB). Le deucravacitinib ne doit pas être administré aux patients présentant une TB active (voir rubrique Contre-indications). Le traitement contre la TB latente doit être mis en place avant d'administrer deucravacitinib. Un traitement antituberculeux doit être envisagé avant d'instaurer un traitement par deucravacitinib chez les patients présentant des antécédents de TB latente ou active, chez lesquels l'administration d'un traitement adéquat ne peut être confirmée. Les patients recevant du deucravacitinib doivent être surveillés pour détecter tout signe et symptôme de TB active.
Tumeurs malignes
Des tumeurs malignes, y compris des lymphomes et des cancers cutanés non mélanomateux (CCNM), ont été observées lors des études cliniques avec deucravacitinib.
Il n'a pas été déterminé si l'inhibition de la tyrosine kinase 2 (TYK2) était possiblement associée aux effets indésirables liés à l'inhibition de Janus kinase (JAK). Dans une étude à grande échelle randomisée, contrôlée par agent actif, menée sur un inhibiteur de JAK chez des patients âgés de 50 ans et plus souffrant de polyarthrite rhumatoïde et présentant au moins un facteur de risque cardiovasculaire supplémentaire, un taux plus élevé de tumeurs malignes, en particulier des tumeurs malignes du poumon, des lymphomes et des CCNM, a été observé avec un inhibiteur de JAK par rapport à des inhibiteurs du facteur de nécrose tumorale (TNF).
Des données cliniques limitées sont disponibles pour évaluer le lien éventuel entre l'exposition au deucravacitinib et le développement de tumeurs malignes. Une évaluation de la sécurité à long terme est en cours. Les risques et bénéfices d'un traitement par deucravacitinib doivent être pris en compte avant d'instaurer le traitement chez les patients.
Événements indésirables cardiovasculaires majeurs (EICM), thrombose veineuse profonde (TVP) etembolie pulmonaire (EP)
Il n'a pas été déterminé si l'inhibition de la TYK2 était possiblement associée aux effets indésirables liés à l'inhibition des JAK. Dans une étude à grande échelle randomisée, contrôlée par agent actif, menée sur un inhibiteur de JAK chez des patients âgés de 50 ans et plus souffrant de polyarthrite rhumatoïde et présentant au moins un facteur de risque cardiovasculaire supplémentaire, un taux plus élevé d'EICM, correspondant à des morts cardiaques, des infarctus du myocarde d'issue non fatale et des accidents cérébrovasculaires d'issue non fatale, et un taux plus élevé de thromboembolie veineuse, notamment de TVP et d'EP, en corrélation avec la dose administrée, ont été observés avec un inhibiteur de JAK par rapport à des inhibiteurs du TNF.
Aucune augmentation du risque d'EICM, de TVP et d'EP n'a été observée dans les études cliniques avec le deucravacitinib. Des évaluations de la tolérance à long terme de deucravacitinib sont en cours. Les risques et bénéfices d'un traitement par deucravacitinib doivent être pris en compte avant d'instaurer le traitement chez les patients.
Vaccinations
Avant
de débuter un traitement par deucravacitinib, il convient d'envisager
la réalisation de toutes les vaccinations appropriées selon l'âge,
conformément aux recommandations vaccinales en vigueur.
L'utilisation
de vaccins vivants atténués doit être évitée chez les patients traités
par deucravacitinib. La réponse aux vaccins vivants atténués et
inactivés n'a pas été évaluée.
Excipients
LactoseCe médicament contient du lactose. Les patients présentant une intolérance au galactose, un déficit total en lactase ou un syndrome de malabsorption du glucose et du galactose (maladies héréditaires rares) ne doivent pas prendre ce médicament.
Sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par comprimé, c.-à-d. qu'il est essentiellement « sans sodium ».
Résumé du profil de sécurité
L'effet indésirable le plus fréquemment rapporté est les infections des voies aériennes supérieures (18,9 %), le plus souvent une rhinopharyngite. Le profil de sécurité du deucravacitinib à plus long terme était similaire et cohérent avec les études de développement.
Liste tabulée des effets indésirables
La liste des effets indésirables suivante pour deucravacitinib provient d'études cliniques sur le psoriasis en plaques (Tableau 1). Ces effets sont présentés par classe de systèmes d'organes MedDRA et par fréquence.
Les fréquences sont définies comme suit : très fréquent (≥ 1/10) ; fréquent (≥ 1/100, < 1/10) ; peu fréquent (≥ 1/1 000, < 1/100) ; rare (≥ 1/10 000, < 1/1 000) ; très rare (< 1/10 000) ; fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Tableau 1 : Liste des effets indésirables
| Classe de systèmes d'organes | Fréquence | Effet indésirable |
| Infections et infestations | Très fréquent | Infections des voies aériennes supérieuresa |
| Fréquent | Infections par le virus de l'herpèsb | |
| Peu fréquent | Zona | |
| Affections gastro-intestinales | Fréquent | Ulcères buccauxc |
| Affections de la peau et du tissu sous-cutané | Fréquent | Rash acnéiformed Folliculite |
| Investigations | Fréquent | Créatine phosphokinase sanguine augmentée |
| a
Les infections des voies aériennes supérieures comprennent la
rhinopharyngite, l'infection des voies aériennes supérieures,
l'infection virale des voies aériennes supérieures, la pharyngite, la
sinusite, la sinusite aiguë, la rhinite, l'amygdalite, l'abcès
périamygdalien, la laryngite, la trachéite et la rhinotrachéite. b Les infections par le virus de l'herpès comprennent l'herpès buccal, herpes simplex, l'herpès génital et l'infection virale par le virus de l'herpès. c Les ulcères buccaux comprennent l'ulcère aphteux, l'ulcération buccale, l'ulcération linguale et la stomatite. d Le rash acnéiforme comprend l'acné, la dermatite acnéiforme, le rash, la rosacée, les pustules, le rash pustuleux et les papules. | ||
Description de certains effets indésirables
Infections
Dans les études POETYK PSO-1 et POETYK PSO-2 (voir rubrique Propriétés pharmacodynamiques), des infections sont survenues chez 29,1 % des patients traités par deucravacitinib (116,0 événements pour 100 personnes-années) contre 21,5 % des patients traités par le placebo (83,7 événements pour 100 personnes-années) au cours des 16 premières semaines. La majorité des infections étaient sans gravité, d'intensité légère à modérée et n'ont pas entraîné d'arrêt du traitement par deucravacitinib. L'incidence des infections graves chez les patients traités par deucravacitinib était de 0,6 % (2,0 événements pour 100 personnes-années) et de 0,5 % chez les patients traités par le placebo (1,6 événement pour 100 personnes-années).
Dans les études POETYK PSO-1 et POETYK PSO-2 (voir rubrique Propriétés pharmacodynamiques), des infections sont survenues chez 29,1 % des patients traités par deucravacitinib (116,0 événements pour 100 personnes-années) contre 21,5 % des patients traités par le placebo (83,7 événements pour 100 personnes-années) au cours des 16 premières semaines. La majorité des infections étaient sans gravité, d'intensité légère à modérée et n'ont pas entraîné d'arrêt du traitement par deucravacitinib. L'incidence des infections graves chez les patients traités par deucravacitinib était de 0,6 % (2,0 événements pour 100 personnes-années) et de 0,5 % chez les patients traités par le placebo (1,6 événement pour 100 personnes-années).
Le taux d'infections chez les patients traités par deucravacitinib n'a pas augmenté jusqu'à la semaine 52 (95,4 événements pour 100 personnes-années). Le taux d'infections graves chez les patients traités par deucravacitinib n'a pas augmenté jusqu'à la semaine 52 (1,7 événement pour 100 personnes-années).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration - voir Annexe V.
AVANT le début de traitement :
- EFFECTUER un test de dépistage de l'infection par la tuberculose.
- EVISAGER la réalisation de toutes les vaccinations appropriées selon
l'âge, conformément aux recommandations vaccinales en vigueur.
SURVEILLANCE du traitement :
- signes ou symptômes évoquant une infection,
- signe et symptôme de tuberculose active.
INFORMER le médecin en cas de symptômes suivants : jambe gonflée douloureuse, douleur thoracique ou essoufflement.
Grossesse
Il existe des données limitées sur l'utilisation du deucravacitinib chez la femme enceinte. Les études effectuées chez l'animal n'ont pas mis en évidence d'effets délétères directs ou indirects sur la reproduction (voir rubrique Données de sécurité préclinique). Par mesure de précaution, il est préférable d'éviter l'utilisation du deucravacitinib pendant la grossesse.
Allaitement
On ne sait pas si deucravacitinib/les métabolites sont excrétés dans le lait maternel.
Les données disponibles chez l'animal ont mis en évidence l'excrétion du deucravacitinib dans le lait (voir rubrique Données de sécurité préclinique).
Un risque pour les nouveau-nés/nourrissons en rapport avec l'allaitement ne peut être exclu. La décision d'interrompre l'allaitement ou d'interrompre/de s'abstenir du traitement avec deucravacitinib doit être prise, en prenant en compte le bénéfice de l'allaitement pour l'enfant au regard du bénéfice du traitement pour la femme.
Fertilité
L'effet du deucravacitinib sur la fertilité humaine n'a pas été évalué. Les études effectuées chez l'animal n'ont pas mis en évidence d'effets délétères directs ou indirects sur la fertilité (voir rubrique Données de sécurité préclinique).
Les études cliniques indiquent que deucravacitinib ne présente pas d'interaction médicamenteuse cliniquement significative en cas d'administration concomitante d'autres médicaments indiqués ci-dessous ; par conséquent, aucune adaptation posologique n'est nécessaire.
Effet du deucravacitinib sur d'autres médicaments
Le deucravacitinib n'a pas d'impact significatif sur les expositions plasmatiques à la rosuvastatine (substrats de la protéine de résistance au cancer du sein [BCRP] et du polypeptide de transport d'anions organiques [OATP]), au méthotrexate (substrat de la BCRP et de transporteurs rénaux), au mycophénolate mofétil (MMF) (substrats de CES1 et CES2) ou aux contraceptifs oraux (acétate de noréthindrone et éthinylestradiol).
Effet d'autres médicaments sur le deucravacitinib
Les médicaments ayant un effet inhibiteur ou inducteur sur les enzymes CYP ou les transporteurs tels que la cyclosporine (inhibiteur double de glycoprotéine P [P-gp]/BCRP), la fluvoxamine (inhibiteur puissant du CYP 1A2), le ritonavir (inducteur modéré du CYP 1A2), le diflunisal (inhibiteur de l'UGT 1A9), la pyriméthamine (inhibiteur de l'OCT1), la famotidine (antagoniste du récepteur H2) ou le rabéprazole (inhibiteur de la pompe à protons) n'affectent pas de manière significative les expositions plasmatiques au deucravacitinib (voir la rubrique Propriétés pharmacocinétiques).
Le traitement doit être initié sous la conduite et la surveillance d'un médecin expérimenté dans le diagnostic et le traitement du psoriasis.
Posologie
La dose recommandée est de 6 mg par voie orale une fois par jour.
En l'absence de signes de bénéfice thérapeutique après 24 semaines, l'arrêt du traitement doit être envisagé. La réponse du patient au traitement doit être évaluée régulièrement.
Populations particulières
Personnes âgées
Aucune adaptation posologique n'est nécessaire chez les patients âgés de 65 ans et plus (voir rubrique Propriétés pharmacocinétiques). L'expérience clinique chez les patients de 75 ans ou plus est très limitée et le deucravacitinib doit être utilisé avec précaution dans ce groupe de patients.
Insuffisance rénale
Aucune adaptation posologique n'est nécessaire chez les patients insuffisants rénaux, y compris chez les patients dialysés présentant une insuffisance rénale terminale (voir rubrique Propriétés pharmacocinétiques).
Insuffisance hépatique
Aucune adaptation posologique n'est nécessaire chez les patients présentant une insuffisance hépatique légère à modérée. Il n'est pas recommandé d'utiliser deucravacitinib chez les patients présentant une insuffisance hépatique sévère (voir rubrique Propriétés pharmacocinétiques).
Population pédiatrique
La sécurité d'emploi et l'efficacité du deucravacitinib chez les enfants et les adolescents âgés de moins de 18 ans n'ont pas encore été établies. Aucune donnée n'est disponible.
Aucune adaptation posologique n'est nécessaire chez les patients âgés de 65 ans et plus (voir rubrique Propriétés pharmacocinétiques). L'expérience clinique chez les patients de 75 ans ou plus est très limitée et le deucravacitinib doit être utilisé avec précaution dans ce groupe de patients.
Insuffisance rénale
Aucune adaptation posologique n'est nécessaire chez les patients insuffisants rénaux, y compris chez les patients dialysés présentant une insuffisance rénale terminale (voir rubrique Propriétés pharmacocinétiques).
Insuffisance hépatique
Aucune adaptation posologique n'est nécessaire chez les patients présentant une insuffisance hépatique légère à modérée. Il n'est pas recommandé d'utiliser deucravacitinib chez les patients présentant une insuffisance hépatique sévère (voir rubrique Propriétés pharmacocinétiques).
Population pédiatrique
La sécurité d'emploi et l'efficacité du deucravacitinib chez les enfants et les adolescents âgés de moins de 18 ans n'ont pas encore été établies. Aucune donnée n'est disponible.
Mode d'administration
Voie orale.
Les comprimés peuvent être pris pendant ou en dehors des repas. Les comprimés doivent être avalés entiers et ne doivent pas être écrasés, coupés ou mâchés.
Durée de conservation :
3 ans.
Précautions particulières de conservation :
Ce médicament ne nécessite pas de précautions particulières de conservation.
Sans objet.
Le deucravacitinib a été administré chez des volontaires sains sous forme de doses uniques allant jusqu'à 40 mg (> 6 fois la dose recommandée chez l'homme de 6 mg/jour) et sous forme de doses multiples allant jusqu'à 24 mg/jour (12 mg deux fois par jour) pendant 14 jours sans toxicité limitant la dose.
En cas de surdosage, il est recommandé de surveiller la survenue de signes ou symptômes d'effets indésirables et d'initier immédiatement un traitement symptomatique approprié. La dialyse n'élimine pas totalement deucravacitinib de la circulation systémique (voir rubrique Propriétés pharmacocinétiques).
Classe pharmacothérapeutique : immunosuppresseurs, Code ATC : L04AF07
Mécanisme d'action
Le deucravacitinib inhibe de manière sélective l'enzyme TYK2 (la TYK2 appartient à la famille des JAK). Le deucravacitinib se lie au domaine régulateur de la TYK2, stabilisant une interaction inhibitrice entre les domaines régulateur et catalytique de l'enzyme. Cela entraîne une inhibition allostérique de l'activation médiée par le récepteur de la TYK2 et de ses fonctions cellulaires en aval. La TYK2 sert de médiateur dans la voie de signalisation de l'interleukine-23 (IL-23), l'interleukine-12 (IL-12) et des interférons (IFN) de type I, qui sont des cytokines naturellement présentes impliquées dans les réponses inflammatoires et immunitaires. Le deucravacitinib inhibe la libération des cytokines et des chimiokines pro-inflammatoires.
Effets pharmacodynamiques
Chez les patients atteints de psoriasis, le deucravacitinib a permis de réduire l'expression du gène associé au psoriasis sur peau psoriasique, notamment des réductions de l'expression des gènes régulés par les voies IL-23 et IFN de type I. Le deucravacitinib a permis de réduire les taux d'IL-17A, d'IL-19 et de β-défensine respectivement de 47-50 %, 72 % et 81-84 %, après 16 semaines de traitement à raison d'une prise quotidienne.
Efficacité et sécurité cliniques
L'efficacité et la sécurité d'emploi du deucravacitinib ont été évaluées dans deux études cliniques multicentriques, randomisées, en double aveugle, contrôlées par placebo et par aprémilast (POETYK PSO-1 et POETYK PSO-2) chez des patients âgés de 18 ans et plus atteints de psoriasis en plaques modéré à sévère, qui étaient éligibles à un traitement systémique ou une photothérapie. Les patients présentaient une atteinte de la surface corporelle (SC) ≥ 10 %, un score PASI ≥ 12 et une évaluation sPGA ≥ 3 (modérée ou sévère) sur une échelle à 5 points portant sur la sévérité globale de la maladie.
Les études POETYK PSO-1 et POETYK PSO-2 ont évalué un total de 1 686 patients dont 843 ont été randomisés dans le groupe traité par deucravacitinib 6 mg une fois par jour, 422 dans le groupe traité par aprémilast 30 mg deux fois par jour et 421 dans le groupe placebo.
Dans les deux études, les patients recevant le placebo ont été randomisés sous deucravacitinib à la semaine 16 et ont poursuivi jusqu'à la semaine 52. Les patients randomisés dans le groupe aprémilast qui n'ont pas obtenu de réponse PASI 50 (POETYK PSO-1) ou PASI 75 (POETYK PSO-2) à la semaine 24 ont été randomisés sous deucravacitinib et ont poursuivi jusqu'à la semaine 52. Dans l'étude POETYK PSO-1, les patients randomisés dans le groupe traité par deucravacitinib ont poursuivi le traitement jusqu'à la semaine 52. Dans l'étude POETYK PSO-2, les patients traités par deucravacitinib qui ont obtenu un score PASI 75 à la semaine 24 ont été à nouveau randomisés selon un ratio de 1:1 pour poursuivre le traitement par deucravacitinib (entretien) ou sous placebo (arrêt).
Les caractéristiques de la maladie à l'inclusion étaient homogènes pour la population de l'étude dans les deux études : la majorité des patients était des hommes (67 %), l'âge moyen était d'environ 47 ans, la majorité des patients ayant entre 40 et 64 ans. 10 % des patients étaient âgés de 65 ans ou plus. Le score PASI global médian était de 18,7 et l'atteinte de la SC médiane était de 20 %. Le score sPGA à l'inclusion était de 3 (modéré) chez 79,8 % des patients et de 4 (sévère) chez 20,2 % des patients. Le score de l'indice médian de la qualité de vie spécifique aux maladies dermatologiques (Dermatology Life Quality Index, DLQI) était de 11. Au total, 18,4 % des patients de l'étude présentaient des antécédents de rhumatisme psoriasique.
Dans les deux études, 40 % des patients avaient reçu une photothérapie antérieure, 42,4 % étaient naïfs de tout traitement systémique (y compris les traitements biologiques et/ou non biologiques), 41 % avaient reçu un traitement systémique non biologique antérieur et 34,8 % avaient reçu un traitement biologique antérieur (16,1 % des inhibiteurs de TNF, 4,9 % des inhibiteurs de l'IL-12/23, 16,6 % des inhibiteurs de l'IL-17 et 4,4 % des inhibiteurs de l'IL-23).
Les critères d'évaluation principaux dans les deux études étaient les proportions de patients ayant obtenu 1) une amélioration d'au moins 75 % du score PASI (PASI 75) depuis l'inclusion et 2) un score sPGA « blanchi » ou quasi « blanchi » (0 ou 1) à la semaine 16 comparativement au placebo.
Dans l'étude POETYK PSO-1, 58,4 % des patients traités par deucravacitinib ont obtenu un score PASI 75 à la semaine 16, contre 35,1 % des patients traités par aprémilast et 12,7 % des patients recevant le placebo. À la semaine 16, 53,6 %, 32,1 % et 7,2 % des patients des groupes deucravacitinib, aprémilast et placebo, respectivement, ont obtenu un score sPGA « blanchi » ou quasi« blanchi ». Le deucravacitinib s'est montré supérieur au placebo sur ces critères d'évaluation principaux. Des résultats cohérents ont été observés dans l'étude POETYK PSO-2.
Le tableau 2 présente les principaux résultats d'efficacité pour les critères d'évaluation principaux et les autres critères.
Tableau 2 : Principaux résultats relatifs à l'efficacité chez les adultes atteints de psoriasis en plaques
| POETYK PSO-1 | POETYK PSO-2 | |||||
| Critère d'évaluation | Deucravacitini b (N = 332) n (%) | Aprémilast (N = 168) n (%) | Placebo (N = 166) n (%) | Deucravacitinib (N = 511) n (%) | Aprémilast (N = 254) n (%) | Placebo (N = 255) n (%) |
| sPGA 0/1 | ||||||
| Semaine 16 | 178 (53,6) | 54 (32,1)d | 12 (7,2)a,d | 253 (49,5) | 86 (33,9)d | 22 (8,6)a,d |
| Semaine 24 | 195 (58,7) | 52 (31,0)d | - | 251 (49,8)b | 75 (29,5)d | - |
| sPGA 0 | ||||||
| Semaine 16 | 58 (17,5) | 8 (4,8)d | 1 (0,6)d | 80 (15,7) | 16 (6,3)e | 3 (1,2)d |
| PASI 75 | ||||||
| Semaine 16 | 194 (58,4) | 59 (35,1)d | 21 (12,7)a,d | 271 (53,0) | 101 (39,8)e | 24 (9,4)a,d |
| Semaine 24 | 230 (69,3) | 64 (38,1)d | - | 296 (58,7)b | 96 (37,8)d | - |
| PASI 90 | ||||||
| Semaine 16 | 118 (35,5) | 33 (19,6)e | 7 (4,2)d | 138 (27,0) | 46 (18,1)f | 7 (2,7)d |
| Semaine 24 | 140 (42,2) | 37 (22,0)d | - | 164 (32,5)b | 50 (19,7)d | - |
| PASI 100 | ||||||
| Semaine 16 | 47 (14,2) | 5 (3,0)d | 1 (0,6)d | 52 (10,2) | 11 (4,3)f | 3 (1,2)d |
| PGA 0/1spécifiqueau cuir cheveluc | (N = 209) | (N = 110) | (N = 121) | (N = 305) | (N = 166) | (N = 173) |
| Semaine 16 | 147 (70,3) | 43 (39,1)d | 21 (17,4)d | 182 (59,7) | 61 (36,7)d | 30 (17,3)d |
| L'imputation des non-répondeurs (NRI) a été utilisée ; les patients ayant arrêté le traitement ou quitté l'étude avant que le critère d'évaluation puisse être mesuré ou pour lesquels il manquait des données ont été comptés comme non-répondeurs. a Co-critère d'évaluation principal comparant deucravacitinib au placebo b N = 504 correspondant aux évaluations manquées en raison de la pandémie de COVID-19 c Comprend les patients présentant un score PGA spécifique au cuir chevelu ≥ 3 à l'inclusion d p ≤ 0,0001 pour comparaison entre deucravacitinib et le placebo ou deucravacitinib et l'aprémilast e p < 0,001 pour comparaison entre deucravacitinib et l'aprémilast f p < 0,01 pour comparaison entre deucravacitinib et l'aprémilast | ||||||
L'évaluation de l'âge, du sexe, de l'origine ethnique, du poids corporel, de la durée de la maladie, de la sévérité de la maladie à l'inclusion et de l'existence d'un traitement antérieur par agents biologiques ou non biologiques n'a identifié aucune différence de réponse au deucravacitinib parmi ces sous-groupes.
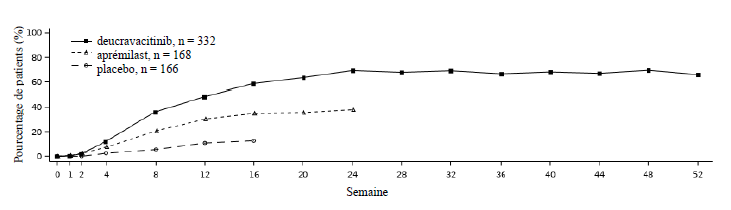
Réponse dans le temps
Le
deucravacitinib a montré une efficacité rapide avec une réponse
maximale PASI 75 obtenue à la semaine 24 (POETYK PSO-1 et PSO-2) et
maintenue jusqu'à la semaine 52 (POETYK PSO-1) (voir Figure 1).
Figure 1 : Réponse (NRI) PASI 75 jusqu'à la semaine 52 par visite dans l'étude POETYK PSO-1
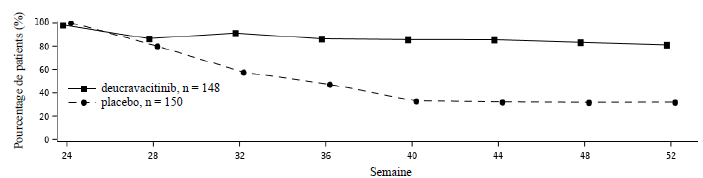
Maintien et durabilité de la réponse
Dans
l'étude POETYK PSO-2, afin d'évaluer le maintien et la durabilité de la
réponse, les patients initialement randomisés dans le groupe
deucravacitinib et ayant obtenu une réponse PASI 75 à la semaine 24 ont
été à nouveau randomisés soit pour poursuivre le traitement par
deucravacitinib soit pour recevoir le placebo. Pour les répondeurs à la
semaine 24 qui ont été à nouveau randomisés dans le groupe sous
placebo, le délai médian avant une perte de réponse PASI 75 était
d'environ 12 semaines. La figure 2 montre les réponses PASI 75 dans les deux bras de la semaine 24 à la semaine 52.
Figure 2 : Réponse (NRI) PASI 75 après une nouvelle randomisation à la semaine 24 dans l'étude POETYK PSO-2
Résultats rapportés par les patients
Des
améliorations significativement plus importantes de la qualité de vie
liée à la santé telle que mesurée par l'indice médian de la qualité de
vie spécifique aux maladies dermatologiques (DLQI) et des symptômes
(démangeaisons, douleur, sensation de brûlure et de picotements, et
tiraillement de la peau) et signes de psoriasis (sécheresse cutanée,
gerçures, desquamation, exfoliation cutanée, rougeur et saignement)
rapportés par les patients tels que mesurés par le journal des signes
et symptômes du psoriasis (Psoriasis Symptoms and Signs Diary, PSSD)
ont été observées chez les patients traités par deucravacitinib par
rapport au placebo à la semaine 16 et par rapport à l'aprémilast à la
semaine 16 et à la semaine 24. L'amélioration de ces réponses chez les
patients recevant un traitement continu par deucravacitinib a été
maintenue jusqu'à la semaine 52 dans l'étude POETYK PSO-1.
Tableau 3 : Résultats rapportés par les patients dans les études POETYK PSO-1 et POETYK PSO-2
| POETYK PSO-1 | POETYK PSO-2 | |||||
| Deucravacitinib | Aprémilast | Placebo | Deucravacitinib | Aprémilast | Placebo | |
| DLQI Patients obtenant 0 ou 1 (NRI)* | N = 322 | N = 161 | N = 160 | N = 495 | N = 247 | N = 246 |
| Semaine 16, n (%) | 132 (41,0) | 46 (28,6)a | 17 (10,6)b | 186 (37,6) | 57 (23,1)b | 24 (9,8)b |
| Semaine 24, n (%) | 155 (48,1) | 39 (24,2)b | - | 205 (41,4) | 53 (21,5)b | - |
| Score des symptômes selon le PSSD Variation depuisl'inclusion (mBOCF)** | N = 306 | N = 158 | N = 151 | N = 466 | N = 233 | N = 239 |
| Semaine 16, moyenne (ET) | -26,7 (1,8) | -17,8 (2,2)b | -3,6 (2,1)b | -28,3 (1,1) | -21,1 (1,4)b | -4,7 (1,4)b |
| Semaine 24, moyenne (ET) | -31,9 (2,0) | -20,7 (2,4)b | - | -29,1 (1,1) | -21,4 (1,5)b | - |
| Score des signes selon le PSSD Variation depuis l'inclusion (mBOCF)* | N = 306 | N = 158 | N = 151 | N = 466 | N = 233 | N = 239 |
| Semaine 16, moyenne (ET) | -28,9 (1,8) | -20,0 (2,2)b | -5,3 (2,1)a | -31,9 (1) | -23,8 (1,4)b | -7,1 (1,4)b |
| Semaine 24, moyenne (ET) | -33,8 (2,0) | -22,5 (2,4)b | - | -32,4 (1,1) | -24,2 (1,5)b | - |
| * Patients présentant un score à l'inclusion ≥ 2 ** Variation moyenne ajustée ; version modifiée du report de la valeur observé à l'inclusion (mBOCF) ; écart-type (ET) a p < 0,01 pour comparaison entre deucravacitinib et placebo ou deucravacitini b et aprémilastb p < 0,0001 pour comparaison entre deucravacitinib et placebo ou deucravacitinib et aprémilast | ||||||
Personnes âgées
Sur les 1 519 patients atteints de psoriasis en plaques traités par deucravacitinib dans les études cliniques, 152 patients étaient âgés de 65 ans ou plus, dont 21 patients de 75 ans ou plus (voir rubrique Posologie et mode d'administration). Aucune différence globale concernant l'exposition, la sécurité ou l'efficacité n'a été observée entre les patients plus âgés et plus jeunes ayant reçu du deucravacitinib.
Population pédiatrique
L'Agence européenne des médicaments a différé l'obligation de soumettre les résultats d'études réalisées avec SOTYKTU dans un ou plusieurs sous-groupes de la population pédiatrique pour le traitement du psoriasis (voir rubrique Posologie et mode d'administration pour les informations concernant l'usage pédiatrique).
Le deucravacitinib a présenté une absorption orale presque complète et une augmentation de l'exposition liée à la dose, et sa pharmacocinétique ne semble pas être dépendante du temps.
Absorption
Suite à l'administration orale de comprimés, deucravacitinib a présenté une absorption rapide et presque complète. La Tmax médiane allait de 2 à 3 heures et la biodisponibilité absolue par voie orale était de 99 % chez les volontaires sains. Une faible accumulation (< 1,4 fois à l'état d'équilibre) a été observée après l'administration une fois par jour.
Alimentation
Le deucravacitinib peut être administré sans tenir compte des aliments ou des anti-sécrétoires gastriques (antagonistes du récepteur H2 et inhibiteurs de la pompe à protons). La prise concomitante
d'aliments ou de anti-sécrétoires gastriques n'a pas affecté l'exposition totale (ASC[INF]) du deucravacitinib.
Distribution
Le volume de distribution à l'état d'équilibre (Vss) est de 140 L, ce qui est supérieur au volume total d'eau dans le corps (42 L), indiquant une distribution extravasculaire. Le deucravacitinib est lié à 81,6 % aux protéines plasmatiques humaines, principalement à l'albumine sérique humaine.
Le deucravacitinib se répartit de façon similaire entre les composants plasmatiques et érythrocytaires, avec un rapport de concentration sang/plasma de 1,26.
Biotransformation
Chez l'homme, deucravacitinib est métabolisé via quatre voies principales de biotransformation, qui comprennent la N-déméthylation au niveau du fragment de triazole par le cytochrome P-450 (CYP) 1A2 pour former le métabolite principal BMT-153261, l'hydrolyse du cyclopropyl- carboxamide par la carboxylestérase 2 (CES2) pour former le métabolite principal BMT-158170, la N-glucurono-conjugaison par uridine glucuronosyltransférase (UGT) pour former le BMT-334616 et la mono-oxydation par le CYP 2B6/2D6 au niveau du groupe méthyle deutéré pour former M11.
À l'état d'équilibre, deucravacitinib est la principale forme circulante constituant 49 % des composants mesurés liés au composé. Deux principaux métabolites circulants, BMT-153261 et BMT-158170, ont été identifiés ; ils présentent tous deux des demi-vies comparables au composant parent, deucravacitinib. BMT-153261 a une efficacité comparable au composant parent et BMT-158170 n'est pas actif sur le plan pharmacologique. L'exposition circulante de BMT-153261 est bien plus faible que celle du composant parent ; par conséquent, la principale activité pharmacologique est attribuée au composant parent, deucravacitinib.
En outre, aucun métabolite propre à l'homme et aucun métabolite circulant à longue durée de vie n'a été identifié.
Élimination
Le deucravacitinib est éliminé par plusieurs voies, y compris le métabolisme de phases I et II, ainsi que l'élimination rénale et fécale directe. En outre, aucune enzyme n'a, à elle seule, contribué à plus de 26 % de la clairance totale. Le deucravacitinib est largement métabolisé, avec 59 % de la dose de [14C]-deucravacitinib administrée par voie orale éliminés sous forme de métabolites dans l'urine (37 % de la dose) et les fèces (22 % de la dose). Le deucravacitinib sous forme inchangée dans l'urine et les fèces représentait 13 % et 26 % de la dose, respectivement.
La demi-vie d'élimination terminale du deucravacitinib 6 mg chez les volontaires sains adultes est de 10 heures, avec une clairance totale de 15,3 L/h (CV 27 %). Le deucravacitinib est un substrat des transporteurs d'efflux, de la glycoprotéine-P (P-gp) et de la protéine de résistance au cancer du sein (BCRP), et du transporteur d'influx OCT1. En raison de la perméabilité passive élevée, de la biodisponibilité orale élevée et de la faible affinité pour ces transporteurs, la contribution de ces derniers à la pharmacocinétique du deucravacitinib est minime.
Le deucravacitinib n'est pas un substrat des transporteurs OATP, NTCP, OAT1, OAT3, OCT2, MATE1 ou MATE2K.
Linéarité/non-linéarité
La pharmacocinétique des doses uniques de deucravacitinib administrées sous forme de comprimés était linéaire pour l'intervalle de doses allant de 3 mg à 36 mg.
Interactions
Effet du deucravacitinib sur d'autres médicaments
Les études in vitro n'ont
mis en évidence aucun effet inhibiteur du deucravacitinib et de ses
principaux métabolites circulants, lors d'expositions cliniques
pertinentes, sur les principaux CYP (1A2, 2B6, 2C8, 2C9, 2C19, 2D6,
3A4), UGT (1A1, 1A4, 1A6, 1A9, 2B7), CES2 et transporteurs de
médicaments (P-gp, BCRP, OATP1B1, OATP1B3, BSEP, MRP2, OAT1, OAT3,
OCT1, OCT2, MATE1 et MATE2K). En outre, deucravacitinib n'induit pas les CYP 1A2, 2B6, et 3A4 (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Populations particulières
Personnes âgées
D'après l'analyse pharmacocinétique de la population, l'exposition moyenne au deucravacitinib à l'état d'équilibre (Cmoy,ss)
était supérieure : de 31 % chez les patients âgés de 65 ans à 74 ans [n
= 87 sur 1 387 (6,3 %)] et de 53 % chez les patients âgés de 75 ans à
84 ans [n = 13 sur 1 387 (0,94 %)]. Les expositions chez les patients
âgés de 85 ans ou plus ne sont pas disponibles.
Patients présentant une insuffisance rénale
Il
n'y a pas eu d'effet cliniquement significatif sur l'exposition au
deucravacitinib chez les patients avec une insuffisance rénale (voir
rubrique Posologie et mode d'administration) d'après une étude
dédiée où le débit de filtration glomérulaire estimé (DFG estimé) était
établi à l'aide de l'équation MDRD (Modification of Diet in Renal
Disease, modification du régime alimentaire en cas de maladie rénale).
Par rapport au groupe présentant une fonction rénale normale, la Cmax du deucravacitinib était modifiée de 15 % maximum et l'ASC[INF]
avait augmenté de 48 % maximum dans les groupes présentant une atteinte
de la fonction rénale (légère [DFG estimé : ≥ 60 à < 90 mL/min],
modérée [DFG estimé : ≥ 30 à < 60 mL/min], sévère [DFG estimé : <
30 mL/min] et terminale [DFG estimé : < 15 mL/min]). Par rapport au
groupe présentant une fonction rénale normale, la Cmax de BMT-153261 avait augmenté de 34 % maximum et l'ASC[INF] avait augmenté de 84 % maximum dans les groupes présentant une atteinte de la fonction rénale.
La dialyse n'élimine pas totalement deucravacitinib de la circulation systémique (5,4 % de la dose est éliminée par dialyse).
Patients présentant une insuffisance hépatique
Il
n'y a pas d'effet cliniquement significatif d'une insuffisance
hépatique légère (classe A selon le score de Child-Pugh) et modérée
(classe B selon le score de Child-Pugh) sur l'exposition au
deucravacitinib chez les patients avec (voir rubrique Posologie et mode d'administration). Comparé au groupe présentant une fonction hépatique normale, la Cmax et l'ASC[INF]
de la fraction totale du deucravacitinib dans le groupe présentant une
insuffisance hépatique légère et modérée avaient augmenté de 10 % et 40
% maximum, respectivement, tandis que la Cmax et l'ASC(INF)
de la fraction libre du deucravacitinib avaient augmenté de 26 % et 60
% maximum, respectivement. Chez les adultes présentant une insuffisance
hépatique sévère (classe C selon le score de Child-Pugh), la Cmax
de la fraction totale du deucravacitinib était comparable tandis que
l'ASC de la fraction totale était supérieure de 43 % par rapport aux
volontaires sains adultes appariés. Chez ces adultes, la Cmax et l'ASC(INF)
de la fraction libre avaient augmenté de 62 % et 131 %, respectivement.
L'utilisation du deucravacitinib n'est pas recommandée chez les
patients présentant une insuffisance hépatique sévère (voir rubrique Posologie et mode d'administration).
L'ASC(0-T) de BMT-153261 avait diminué de 19 %, 53 % et 76 % chez les sujets présentant une insuffisance hépatique légère, modérée et sévère, respectivement, comparé aux sujets présentant une fonction hépatique normale, tandis que la Cmax de BMT-153261 avait diminué de 25 %, 59 % et 79 % chez les sujets présentant une insuffisance hépatique légère, modérée et sévère, respectivement.
Sexe
D'après
la modélisation et la simulation pharmacocinétique de la population, il
est attendu que les femmes aient une exposition moyenne au
deucravacitinib à l'état d'équilibre (Cmax,ss et Cmoy,ss) d'environ 30 % supérieure à celles des hommes.
Poids corporel
D'après
la modélisation et la simulation pharmacocinétique de la population, il
est attendu que les patients dont le poids corporel est plus faible
(< 60 kg) aient une exposition moyenne géométrique au
deucravacitinib à l'état d'équilibre plus élevée : 37,4 % (Cmax,ss) et 24,8 % (Cmoy,ss).
Il est attendu que les patients dont le poids corporel est plus élevé
(> 90 kg) aient une exposition moyenne géométrique au
deucravacitinib à l'état d'équilibre plus faible : 24,8 % (Cmax,ss) et 19,6 % (Cmoy,ss) (par rapport aux patients dont le poids corporel est compris entre 60 et 90 kg).
Facteurs intrinsèques
L'origine raciale et l'origine ethnique n'avaient pas d'effet cliniquement significatif sur l'exposition au deucravacitinib.
Le deucravacitinib n'a aucun effet ou un effet négligeable sur l'aptitude à conduire des véhicules et à utiliser des machines.
Les données non cliniques issues des études conventionnelles de pharmacologie de sécurité, toxicologie en administration répétée, génotoxicité, cancérogenèse, et des fonctions de reproduction et de développement, n'ont pas révélé de risque particulier pour l'homme.
Toxicologie en administration répétée
Dans l'étude de toxicité chronique menée sur des rats, des diminutions des numérations de lymphocytes, des cellules de la moelle osseuse et des cellules lymphoïdes dans les tissus lymphoïdes ont été observées à une exposition (ASC) à la dose minimale ayant un effet observé (DMEO) environ 9 fois supérieure à la dose recommandée chez l'homme (DRH). Ces effets n'étaient pas associés à des signes cliniques d'immunosuppression (par ex., des infections). Des diminutions des numérations plaquettaires et des paramètres de masse des globules rouges ont été observées à une exposition (ASC) à la DMEO environ 42 fois supérieure à la DRH. Dans l'étude de toxicité chronique menée sur des singes, des effets cutanés cliniques et microscopiques, ainsi que des diminutions des paramètres de masse des globules rouges ont été observées à une exposition (ASC) à la DMEO environ 7 fois supérieure à la DRH.
Toxicité pour la reproduction et le développement
Le deucravacitinib n'a pas eu d'effet sur la fertilité ou les premiers stades du développement embryonnaire chez les rats et les rates à des expositions (ASC) jusqu'à 247 et 171 fois supérieures environ à la DRH, respectivement.
Le deucravacitinib n'était ni létal pour les embryons ni tératogène à des expositions maternelles (ASC) jusqu'à 266 fois supérieures environ à la DRH chez les rats ou 91/20 (fraction totale/fraction libre) fois la DRH chez les lapins.
Dans une étude portant sur le développement pré et post-natal chez les rats, des poids temporairement plus faibles des ratons ont été observés pendant la période de pré-sevrage à une exposition maternelle (ASC) environ 110 fois supérieure à la DRH. Cet effet a entièrement disparu après la période de sevrage.
Suite à l'administration de deucravacitinib radiomarqué chez des rates allaitantes, deucravacitinib et/ou ses métabolites étaient présents dans le lait avec des rapports de concentration lait/plasma de 2,7 à 30,9.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste I
Prescription réservée aux spécialistes et services DERMATOLOGIE
Prescription réservée aux spécialistes et services MEDECINE INTERNE
Remboursement
en fonction de l'indication (JO du 19/06/2024) :
La
seule indication thérapeutique ouvrant droit à la prise en charge ou au
remboursement par l'assurance maladie est le traitement du psoriasis en plaques modéré à sévère chez les adultes
éligibles à un traitement systémique en cas d'échec, d'intolérance ou de
contre-indication aux médicaments biologiques (anti-TNFa et
anti-interleukines).
Comprimé pelliculé (comprimé)
Comprimé
pelliculé rose, rond, biconvexe, de 8 mm de diamètre, portant les
mentions « BMS 895 » et « 6 mg » gravées sur une face sur deux lignes,
l'autre face étant lisse.
Plaquette transparente en chlorure de polyvinyle/polychlorotrifluoroéthylène (PVC/PCTFE) munie d'un film en aluminium à percer contenant 14 comprimés pelliculés par plaquette (sous forme calendaire).
Présentation : 28 comprimés pelliculés.
Chaque comprimé pelliculé contient 6 mg de deucravacitinib.
Excipient à effet notoire :
Chaque comprimé pelliculé contient 44 mg de lactose (voir rubrique Mises en garde spéciales et précautions d'emploi).
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Noyau du comprimé
Succinate d'acétate d'hypromelloseLactose anhydre
Cellulose microcristalline
Croscarmellose sodique
Silice colloïdale hydratée
Stéarate de magnésium
Enrobage
Alcool polyvinyliqueDioxyde de titane (E171)
Macrogol
Talc
Oxyde de fer rouge (E172)
Oxyde de fer jaune (E172)